From protein accession to mutation-aware structural interpretation.
PDB-MutMap maps protein sequence differences onto experimentally resolved structures, annotates their COSMIC status, and links each mutation to sequence alignment evidence and an interactive Mol* 3D view.
Submit a gene name or UniProt accession
The workflow starts from the home page. Users can submit either a human gene name, such as TP53 or RhoA, or a UniProt accession, such as O75874. After submission, the webserver searches for experimentally resolved PDB structures mapped to the submitted protein sequence.
When mapped PDB structures are available, PDB-MutMap downloads the structural files, extracts the representative protein chain, aligns the PDB chain sequence against the UniProt reference sequence, and generates the result files. If no mapped PDB entry is available, the workflow stops and informs the user.
Interpret the results table
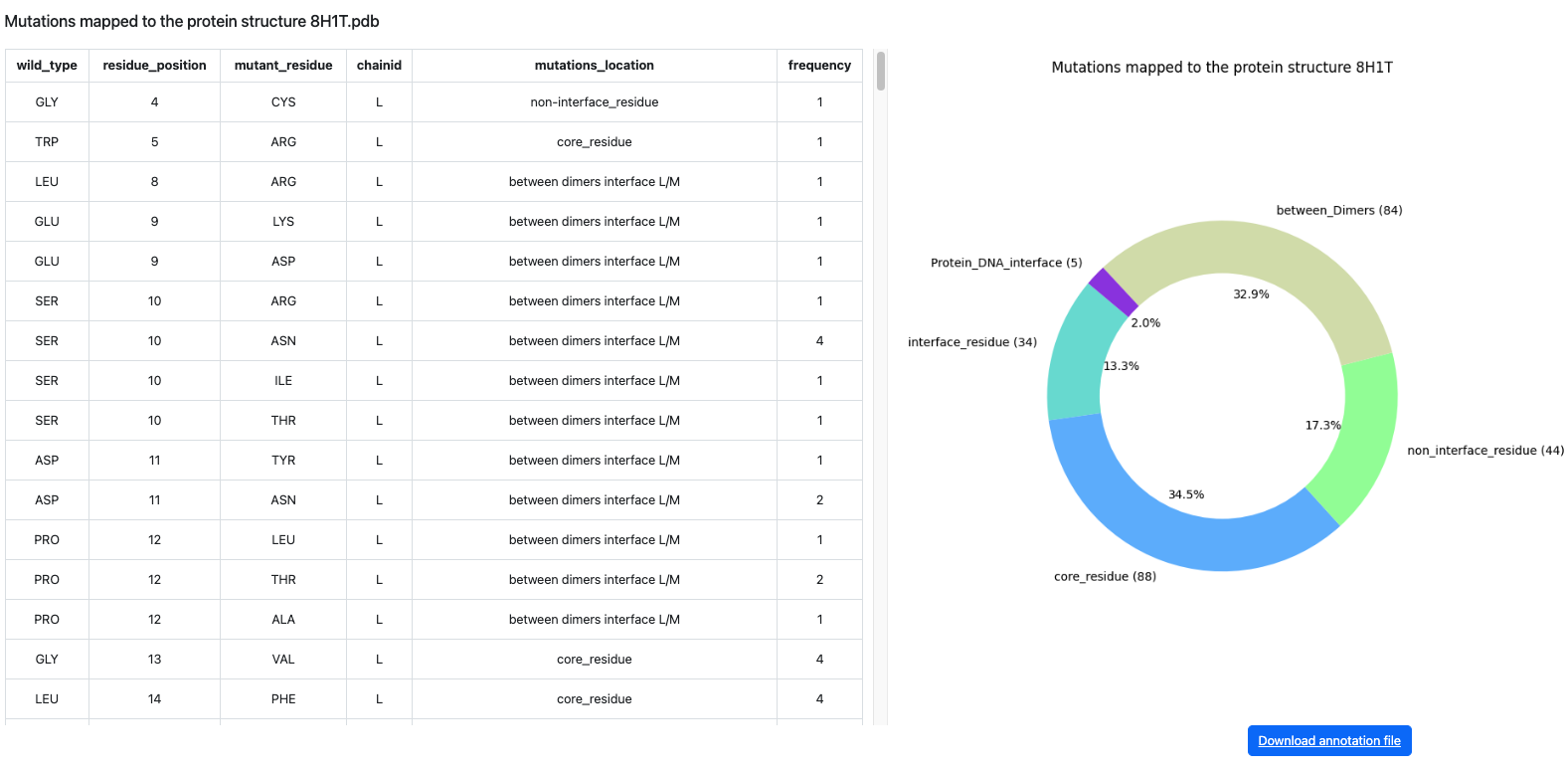
After the analysis is completed, the user is directed to the results table. Each row represents a mapped PDB chain and its detected sequence difference compared with the UniProt reference sequence.
The table helps users rapidly inspect which structures contain mutations, which mutations are reported in COSMIC, and which structures match the reference sequence.
- PDB ID: the experimentally resolved structure mapped to the submitted UniProt protein.
- Chain ID: the representative chain extracted from the PDB file.
- Mutation: the amino-acid difference, for example R132H; fully matched structures are labelled WT.
- Character: classifies the row as in cosmic, not in cosmic, or wild type.
- PDB residue label: the structure residue number used for Mol* localization.
- Identity and assembly: sequence identity, mutation count, chain length, and biological assembly metadata.
The PDB ID is clickable and opens the detailed alignment and structure-viewing page for the selected row.
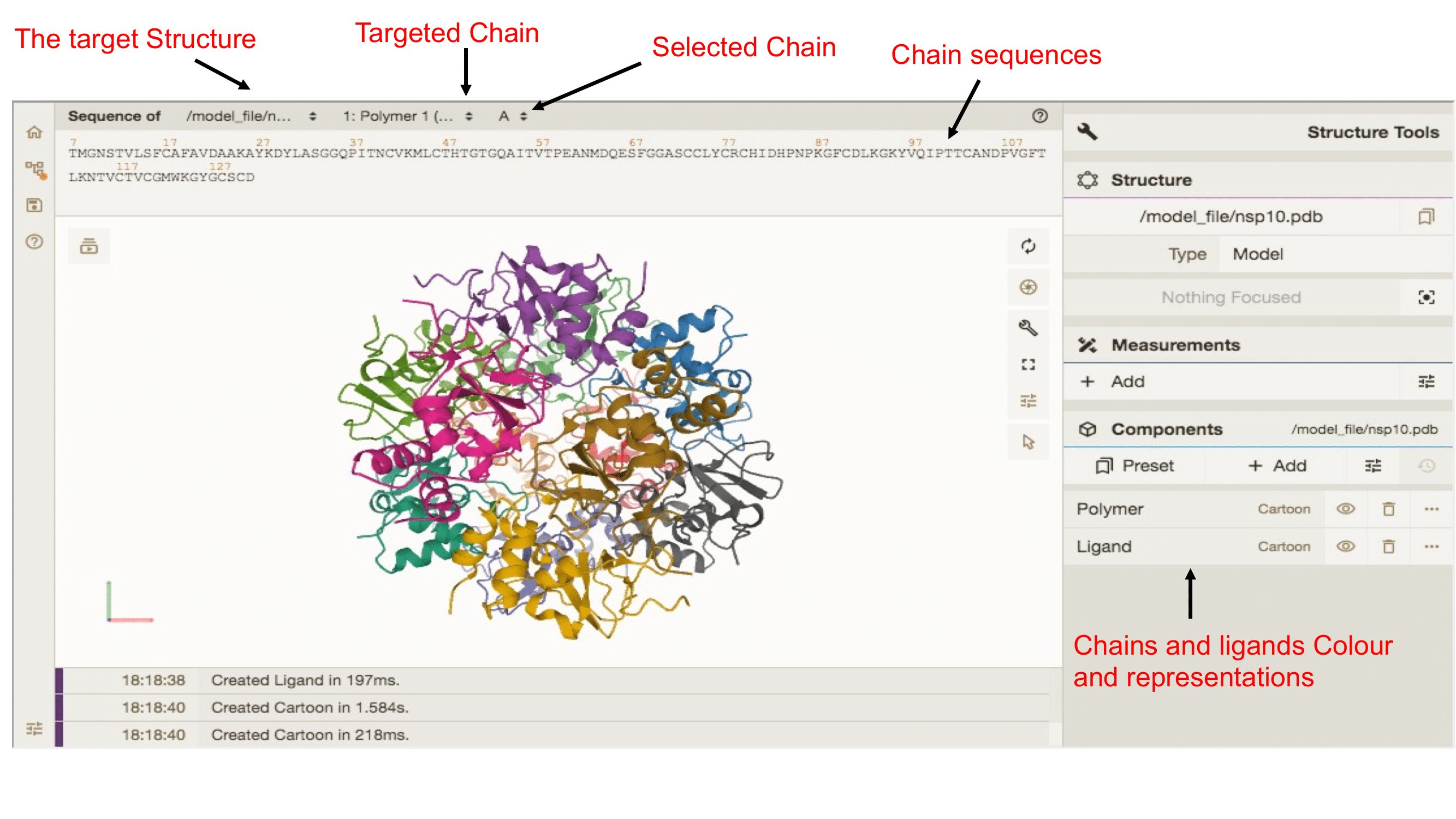
Inspect the selected mutation in Mol
Under the sequence alignment, PDB-MutMap provides an embedded Mol* viewer for interactive inspection of the selected PDB structure. The viewer loads the local PDB file generated by the pipeline and allows users to rotate, zoom, select chains, and inspect residues directly inside the webserver.
When a mutation is selected, PDB-MutMap passes the chain ID and PDB residue label to the Mol* viewer. The selected residue can then be highlighted and focused in the 3D structure, linking the table result to the exact structural position.